MARRVEL

MARRVEL (Model organism Aggregated Resources for Rare Variant ExpLoration) aims to facilitate the use of public genetic resources to prioritize rare human gene variants for study in model organisms. To automate the search process and gather all the data in a simple display we extract data from human data bases (OMIM, ExAC, Geno2MP, DGV, and DECIPHER) for efficient variant prioritization. The protein sequences for six organisms (S. cerevisiae, C. elegans, D. melanogaster, D. rerio, M. musculus, and H. sapiens) are aligned with highlighted protein domain information via collaboration with DIOPT. The key biological and genetic features are then extracted from existing model organism databases (SGD, PomBase, WormBase, FlyBase, ZFIN, and MGI).
SPA-STOCSY: Spatial clustering algorithm – Statistical total correlation spectroscopy
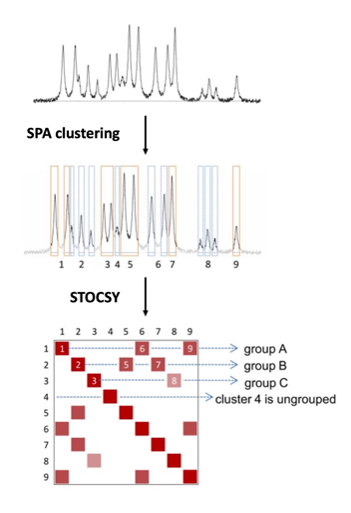
SPA-STOCSY is an automated tool for identifying annotated and non-annotated metabolites in high-throughput NMR spectra. Metabolomics (the study of all metabolites in a given sample) can be a promising candidate for quantitative phenotyping and biomarker discovery. Nuclear magnetic resonance (NMR) is one of the main platforms used to acquire metabolomic data. To accelerate the application of NMR metabolomics in medicine and biology, we present SPA-STOCSY.
SPA exploits strong correlations among datapoints from the multiplets (multiple peaks belonging to the same metabolite) and identifies local spatial clusters of contiguous datapoints highly likely to arise from the same metabolite cluster. With the SPA-derived clusters as input, STOCSY is then used to highlight the correlation map and arrange clusters into highly correlated groups containing signals from the same metabolite. This work has been published in Bioinformatics (https://doi.org/10.1093/bioinformatics/btad593).
CB2
CB2 provides functions for hit gene identification and quantification of sgRNA (single-guided RNA) abundances for CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) pooled screen data analysis.
SalmonTE
SalmonTE is an ultra-Fast and Scalable Quantification Pipeline of Transpose Element (TE) Abundances from Next Generation Sequencing Data. It comes with Salmon which is a fast and accurate transcriptome quantification method.
DASC
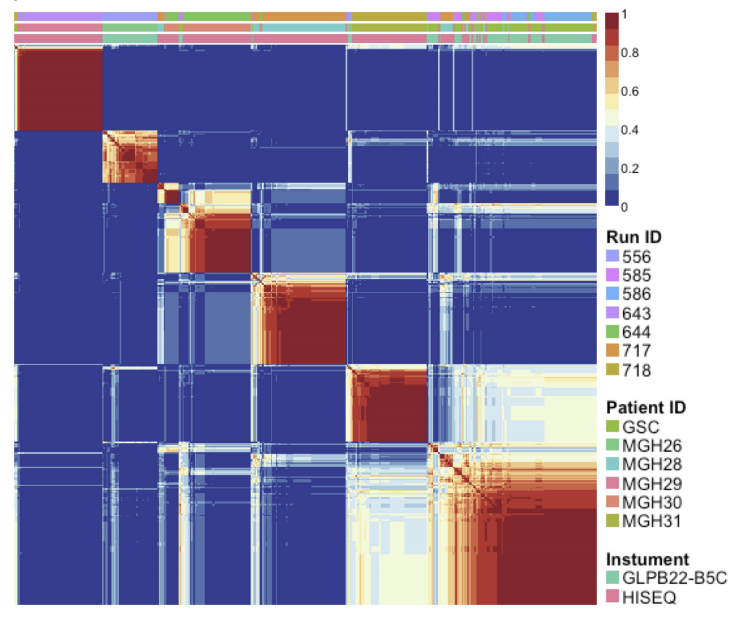
We introduce a new algorithm based on data-adaptive shrinkage and semi-Nonnegative Matrix Factorization (NMF) for the detection of unknown batch effects. We test our algorithm on three different datasets – 1) Sequencing Quality Control (SEQC), 2) Topotecan RNA-Seq and 3) Single-cell RNA-Seq on Glioblastoma Multiforme (GBM). We have demonstrated a superior performance in identifying hidden batch effects as compared to existing algorithms for batch detection in all three datasets. In the Topotecan study, we were able to identify a new batch factor that has been missed by the original study, leading to under-representation of differentially expressed genes. For scRNA-Seq, we demonstrated the power of our method in detecting subtle batch effects.
CRISPRcloud
We present a user-friendly, cloud-based, data analysis pipeline for the deconvolution of pooled screening data. This tool serves a dual purpose of extracting, clustering and analyzing raw next generation sequencing files derived from pooled screening experiments while at the same time presenting them in a user-friendly way on a secure web-based platform. Moreover, CRISPRcloud serves as a useful web-based analysis pipeline for reanalysis of pooled CRISPR screening datasets. Taken together, the framework described in this study is expected to accelerate development of web-based bioinformatics tool for handling all studies which include next generation sequencing data.
CrypSplice
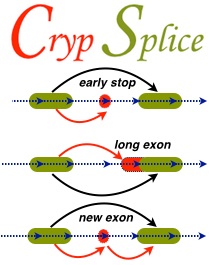
Alternative splicing of RNA is the key mechanism by which a single gene codes for multiple functionally diverse proteins. Recent studies identified previously unknown class of exons, ‘cryptic’ exons, in RNA transcripts. These cryptic exons are often associated with various human cancers and neurological disorders. Genome-wise detection of cryptic splice sites can facilitate a comprehensive understanding of the underlying disease mechanisms and develop strategies that hope to resolve cryptic splicing with the ultimate goal of therapeutic applications. CrypSplic is a novel cryptic splice site detection method. It uses beta-binomial distribution to model junction count data. Every junction is subjected to a beta binomial test w.r.t conditions and classified to aid molecular inferences.
TCGA2STAT: A TCGA data widget for statistical analysis in R.

Large amount of high-throughput data profiled from tumor patients were made publicly available by national projects. However, the process of getting and having these data ready for analyses is intricate for computational researchers, such as statisticians and mathematicians, which hinder them from fully utilizing these abundant resources. We present an open source package, TCGA2STAT, to
obtain TCGA data and prepare the data into format ready for statistical
analysis in R environment. This package can be seamlessly
integrated into computational analyses.
XMRF: An R Package to Fit Markov Networks to High-Throughput Genomics Data

Technological advances in medicine have led to a rapid proliferation of high-throughput “omics” data. Tools to mine this data and discover disrupted disease networks are needed, as they hold the key to understanding complicated interactions between genes, mutations and aberrations, and epi-genetic markers.
Therefore, we developed an R software package, XMRF, that can be used to fit Markov Networks to various types of high-throughput genomics data. Encoding the models and estimation techniques of the recently proposed exponential family Markov Random Fields, our software can be used to learn genetic networks from RNA-sequencing data (counts via Poisson graphical models), mutation and copy number variation data (categorical via Ising models), and methylation data (continuous via Gaussian graphical models).
Combinatorial Therapy Discovery using Mixed Integer Linear Programming
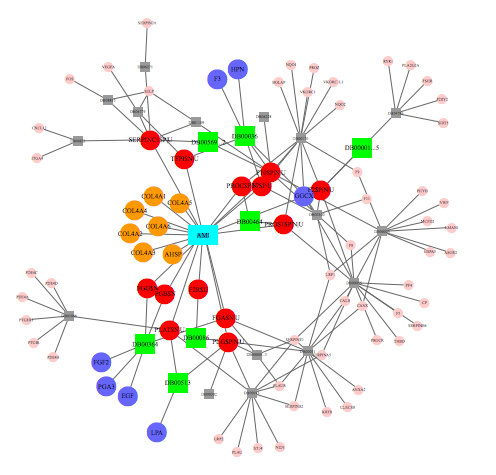
Combinatorial therapies play increasingly important roles in combating complex diseases. Due to the huge cost associated with experimental methods in identifying optimal drug combinations, computational approaches can provide a guide to limit the search space and reduce cost. However, few computational approaches have been developed for this purpose and thus there is a great need of new algorithms for drug combination prediction.
Here we proposed to formulate the optimal combinatorial therapy problem into two complementary mathematical algorithms, Balanced Target Set Cover (BTSC) and Minimum Off-Target Set Cover (MOTSC). Given a disease gene set, BTSC seeks a balanced solution that maximizes the coverage on the disease genes and minimizes the off-target hits at the same time. MOTSC seeks a full coverage on the disease gene set while minimizing the off-target set. Through simulation, both BTSC and MOTSC demonstrated a much faster running time over exhaustive search with the same accuracy. When applied to real disease gene sets, our algorithms not only identified known drug combinations, but also predicted novel drug combinations that are worth further testing. In addition, we developed a web-based tool to allow users to iteratively search for optimal drug combinations given a user-defined gene set.
Logit-Laplacian-net
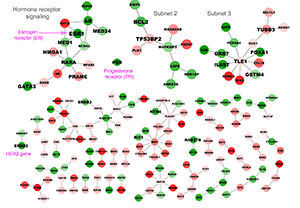
Selecting genes and pathways indicative of disease is a central problem in computational biology. This problem is especially challenging when parsing multi-dimensional genomic data. A number of tools, such as L1-norm based regularization and its extensions elastic net and fused lasso, have been introduced to deal with this challenge. However, these approaches tend to ignore the vast amount of a priori biological network information curated in the literature.
We propose the use of graph Laplacian regularized logistic regression to integrate biological networks into disease classification and pathway association problems. Simulation studies demonstrate that the performance of the proposed algorithm is superior to elastic net and lasso analyses. Utility of this algorithm is also validated by its ability to reliably differentiate breast cancer subtypes using a large breast cancer dataset recently generated by the Cancer Genome Atlas (TCGA) consortium. Many of the protein-protein interaction modules identified by our approach are further supported by evidence published in the literature. Source code of the proposed algorithm is freely available at Github.
DSA: Digital Sorting Algorithm for heterogeneous samples
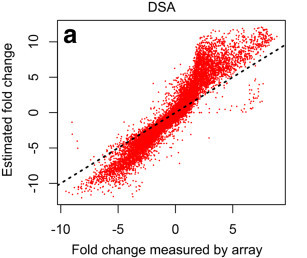
Cellular heterogeneity is present in almost all gene expression profiles. However, transcriptome analysis of tissue specimens often ignores the cellular heterogeneity present in these samples. Standard deconvolution algorithms require prior knowledge of the cell type frequencies within a tissue or their in vitro expression profiles. Furthermore, these algorithms tend to report biased estimations.
Here, we describe a Digital Sorting Algorithm (DSA) for extracting cell-type specific gene expression profiles from mixed tissue samples that is unbiased and does not require prior knowledge of cell type frequencies.
Source code of the proposed algorithm is freely available at Github.
MIST: Missing-value Imputation and in silico region detection for Spatially resolved Transcriptomics
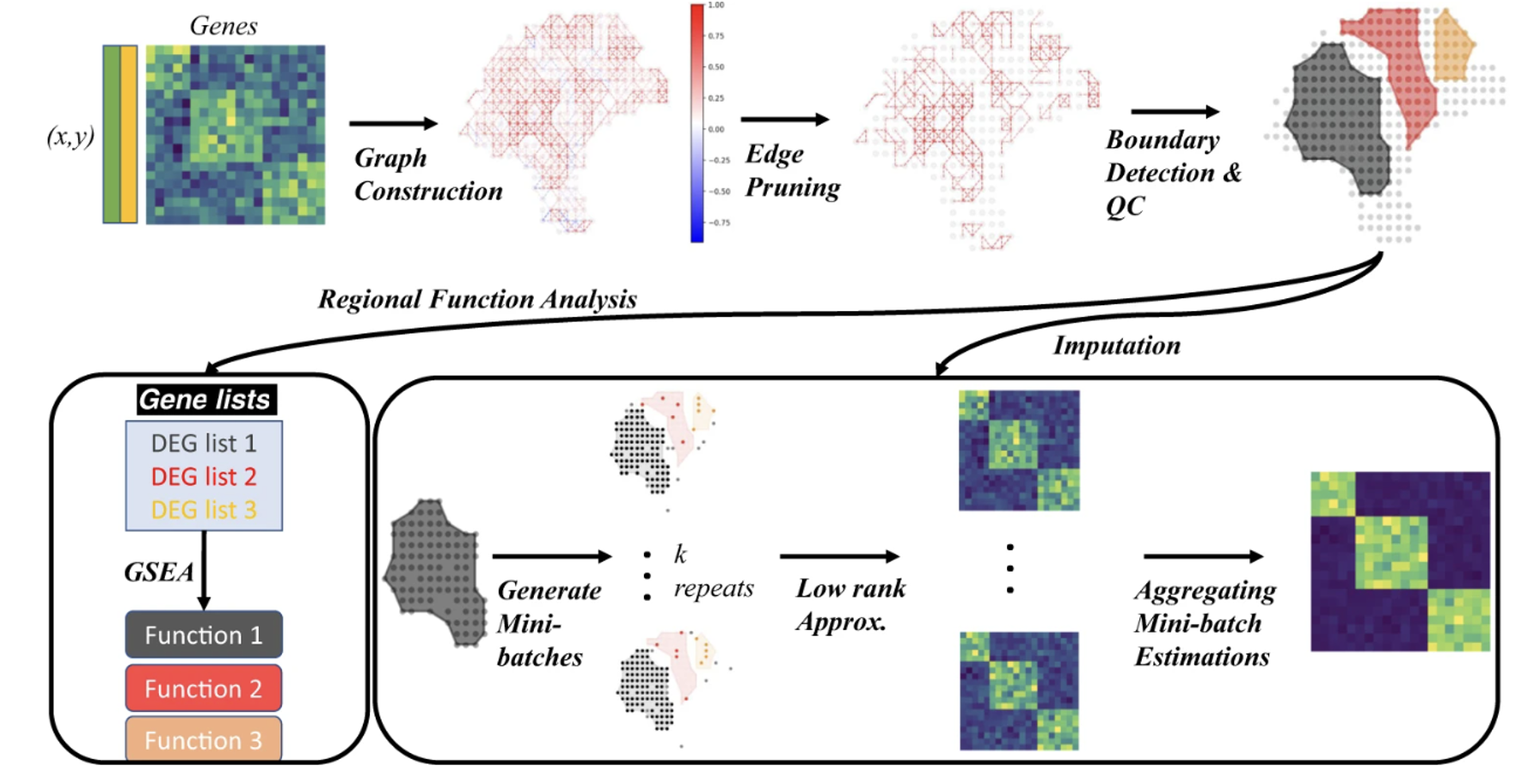
MIST detects tissue regions based on their molecular content by maintaining neighboring spots that are both molecularly similar and physically adjacent. Assuming each detected molecular region has a limited number of cell types, MIST then denoises the missing values by approximating a low-rank gene expression matrix through a nuclear-norm minimization algorithm.
RESORT: Reference-based spatial transcriptomics deconvolution
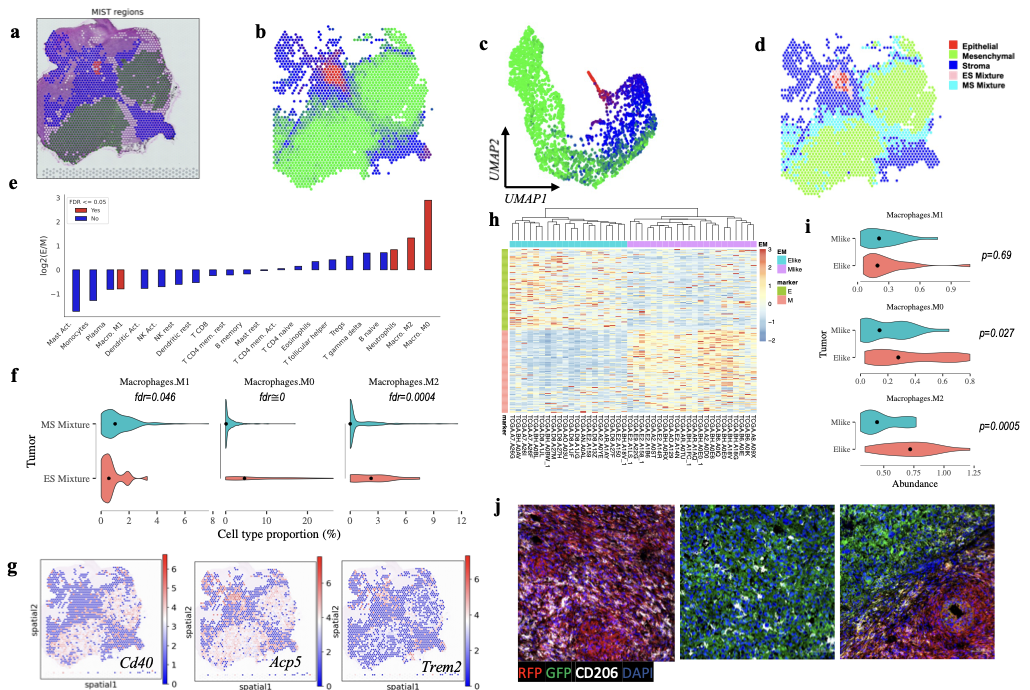
Reference-based spatial transcriptomics deconvolution often faces inaccuracies due to batch/platform discrepancies between references and spatial transcriptomics (ST) data. To tackle this issue, we propose ReSort, a Region-based cell-type Sorting strategy. ReSort extracts spatial regions from ST data, excluding mixture-prone spots, and leverages diverse molecular profiles to create a precise pseudo-internal reference. This approach enables accurate cellular composition estimation in sequencing-based spatial transcriptomics datasets, eliminating the need for external references and potential technical noise.
MinNet: MinNet is a deep learning framework for integrating independent scRNA-seq and scATAC-seq data.
The prototype model ranked 2nd and 4th in NeurIPS 2021 Competition – Open Problem in Single-Cell Analysis, and has been published in BMC Bioinformatics (https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-05126-7). MinNet specializes in batch correction between scRNA-seq and scATAC-seq demonstrated benchmark results, and serves as the first-to-go resource to train tissue-specific integration model.
SEAGAL: Spatial Enrichment Analysis of Gene Associations using L-index
SEAGAL is a Python package for Spatial Enrichment Analysis of Gene Associations using L-index to detect and visualize spatial gene correlations at both single-gene and gene-set levels. Our package takes spatial transcriptomics datasets with gene expression and the aligned spatial coordinates as input. It allows for analyzing and visualizing genes’ spatial correlations and cell types’ colocalization within the precise spatial context. Our manuscript has been published in Bioinformatics (https://academic.oup.com/bioinformatics/article/39/7/btad431/7223197).
TREASMO: Transcription regulation analysis toolkit for single-cell multi-omics data
TREASMO quantifies the single-cell level gene-peak correlation strength, based on which a series of analysis and visualization functions are built to help researchers understand their multi-omics data.
PARMESAN: PARsing ModifiErS via Article aNnotations
PARMESAN is a tool that automatically identifies gene-gene and drug-gene relationships described in PubMed and PubMed Central. It then feeds these extracted relationships into a pathway analysis to predict previously unreported relationships. Each relationship is given a confidence score based on the amount and the consistency of the supporting evidence. Higher confidence scores have been shown to correspond to greater consistency with manually curated molecular pathway databases.
This tool predicts potential genetic and chemical modulators of over 18,000 human genes.
NMRQNet
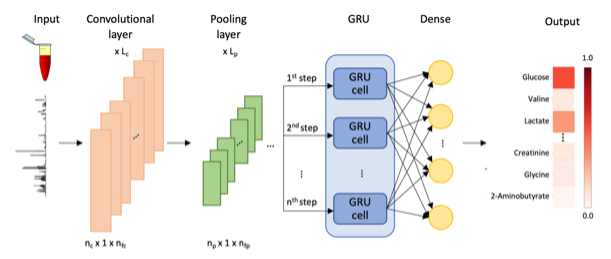
Nuclear magnetic resonance (NMR) is a powerful platform that reveals the metabolomic profile of biofluids or tissues and holds great promise for precision medicine. However, its clinical application has been hindered by the volume and complexity of NMR spectra data as well as the lack of fast and accurate computational tools that can automatically identify and quantify essential metabolites. We present NMRQNet, a deep-learning-based pipeline that can quickly, automatically identify and quantify dominant metabolite candidates within human plasma samples.
NMRQNet takes advantage of CNN, with its achievements in image classification, highlights the spectral features that best separate different metabolites. GRU, with its signal processing abilities, uses the CNN-extracted signals to learn the positional dependencies for clusters belonging to the same metabolite. By combining these two model architectures, the CRNN framework (CNN and GRU) efficiently captures the dominant metabolites.