2021
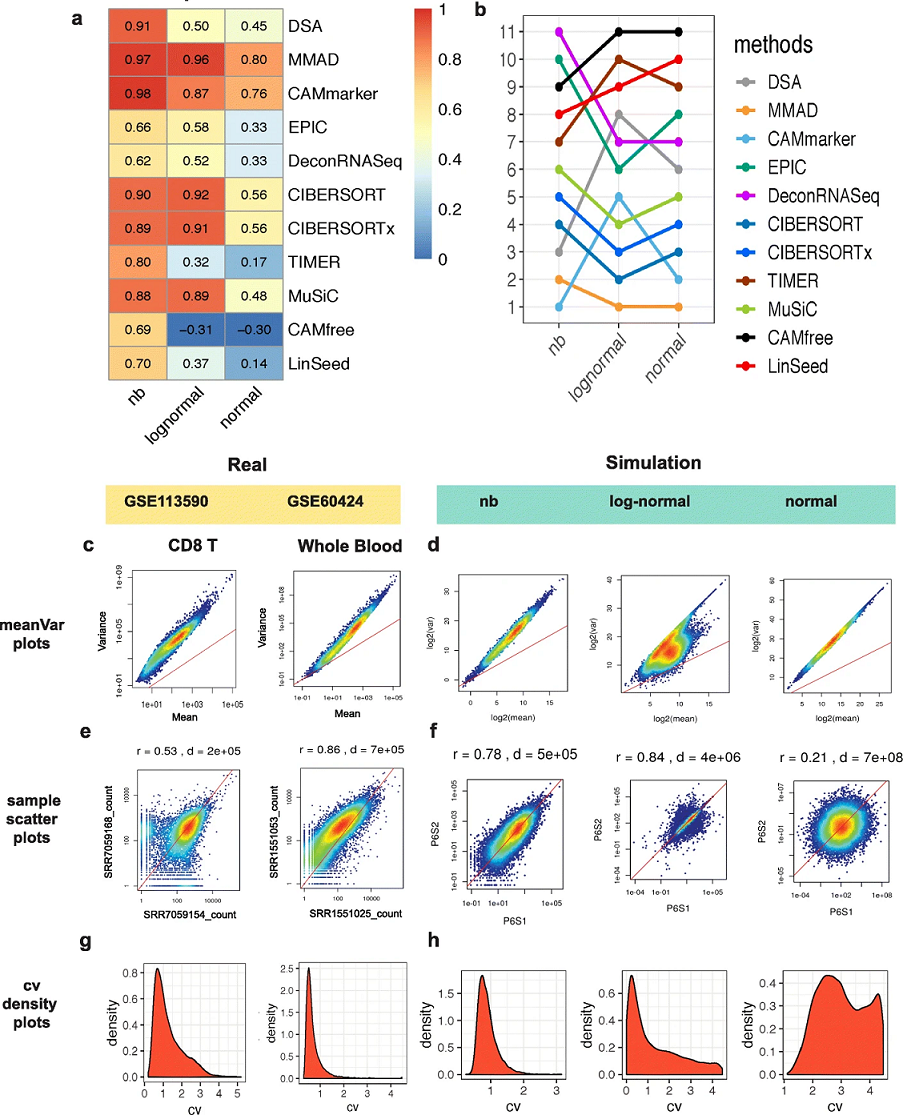
Haijing Jin, Zhandong Liu. “A benchmark for RNA-seq deconvolution analysis under dynamic testing environments.” Genome Biology. doi : 10.1186/s13059-021-02290-6
To systematically reveal the pitfalls and challenges of deconvolution analyses, we investigate the impact of several technical and biological factors including simulation model, quantification unit, component number, weight matrix, and unknown content by constructing three benchmarking frameworks. These frameworks cover comparative analysis of 11 popular deconvolution methods under 1766 conditions. We provide new insights to researchers for future application, standardization, and development of deconvolution tools on RNA-seq data.
2020
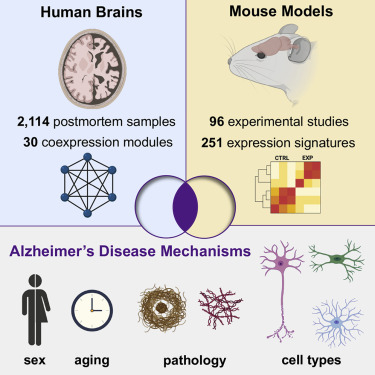
Ying-Wooi Wan, Rami Al-Ouran, et al.“Meta-Analysis of the Alzheimer’s Disease Human Brain Transcriptome and Functional Dissection in Mouse Models.” Cell Reports. doi: 10.1016/j.celrep.2020.107908
We present a consensus atlas of the human brain transcriptome in Alzheimer’s disease (AD), based on meta-analysis of differential gene expression in 2,114 postmortem samples. We discover 30 brain coexpression modules from seven regions as the major source of AD transcriptional perturbations. We next examine overlap with 251 brain differentially expressed gene sets from mouse models of AD and other neurodegenerative disorders. Our results comprise a cross-species resource, highlighting transcriptional networks altered by human brain pathophysiology and identifying correspondences with mouse models for AD preclinical studies.
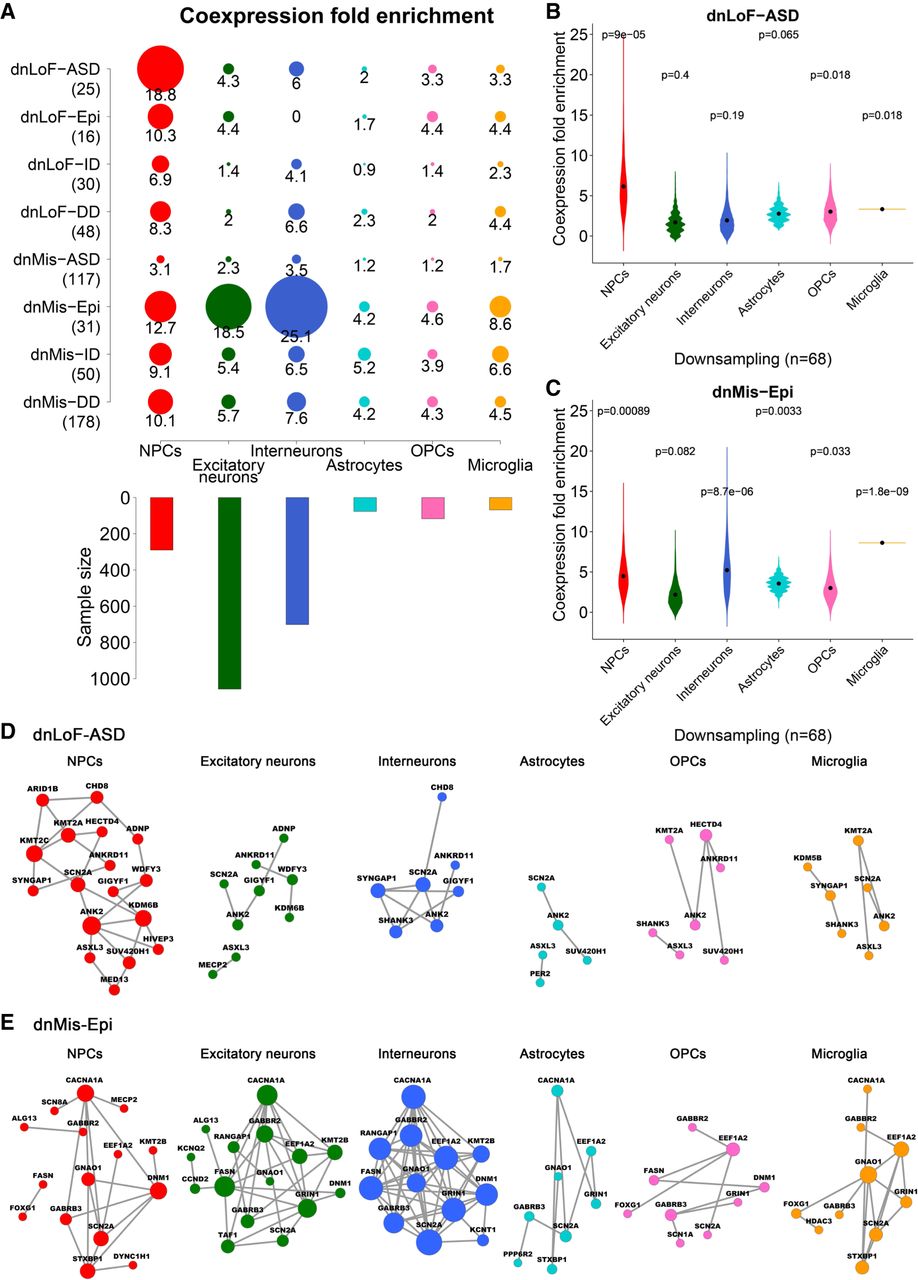
Kaifang Pang, Li Wang, Wei Wang, Jian Zhou, Chao Cheng, Kihoon Han, Huda Y. Zoghbi, Zhandong Liu. “Co-expression enrichment analysis at the single-cell level reveals convergent defects in neural progenitor cells and their cell-type transitions in neurodevelopmental disorders.” Genome Research. doi: 10.1101/gr.254987.119
A large number of genes have been implicated in neurodevelopmental disorders (NDDs), but their contributions to NDD pathology are difficult to decipher without understanding their diverse roles in different brain cell types. Here, we integrated NDD genetics with single-cell RNA sequencing data to assess coexpression enrichment patterns of various NDD gene sets. We identified midfetal cortical neural progenitor cell development—more specifically, the ventricular radial glia-to-intermediate progenitor cell transition at gestational week 10—as a key point of convergence in autism spectrum disorder (ASD) and epilepsy.
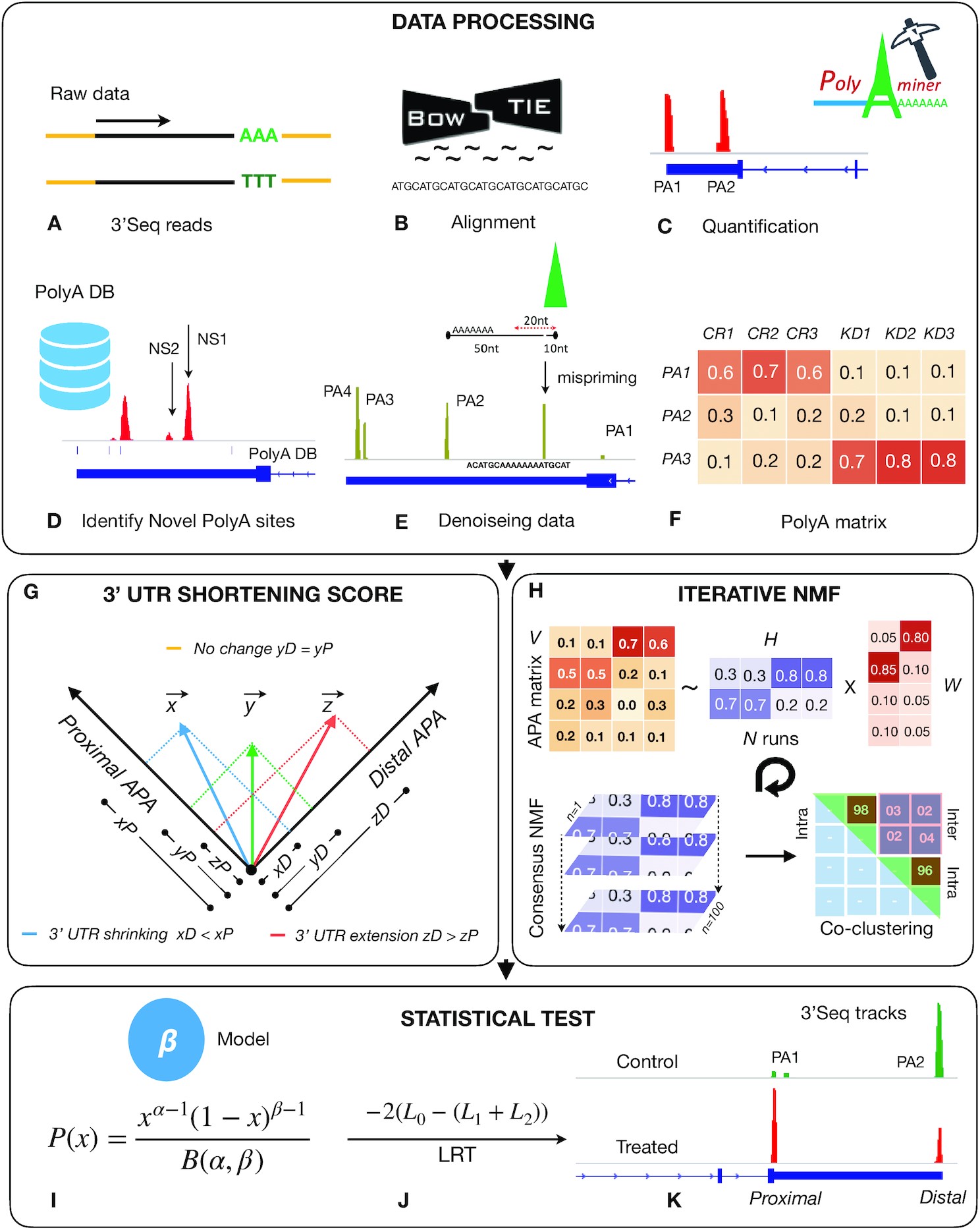
Hari Krishna Yalamanchili, Callison E Alcott, Ping Ji, Eric J Wagner, Huda Y Zoghbi, Zhandong Liu. “PolyA-miner: accurate assessment of differential alternative poly-adenylation from 3′ Seq data using vector projections and non-negative matrix factorization.” Nucleic Acid Research. doi: 10.1093/nar/gkaa398
Almost 70% of human genes undergo alternative polyadenylation (APA) and generate mRNA transcripts with varying lengths, typically of the 3′ untranslated regions (UTR). PolyA-miner can significantly improve data analysis and help decode the underlying APA dynamics. PolyA-miner accounts for all non-proximal to non-distal APA switches using vector projections and reflects precise gene-level 3′UTR changes. It can also effectively identify novel APA sites that are otherwise undetected when using reference-based approaches.
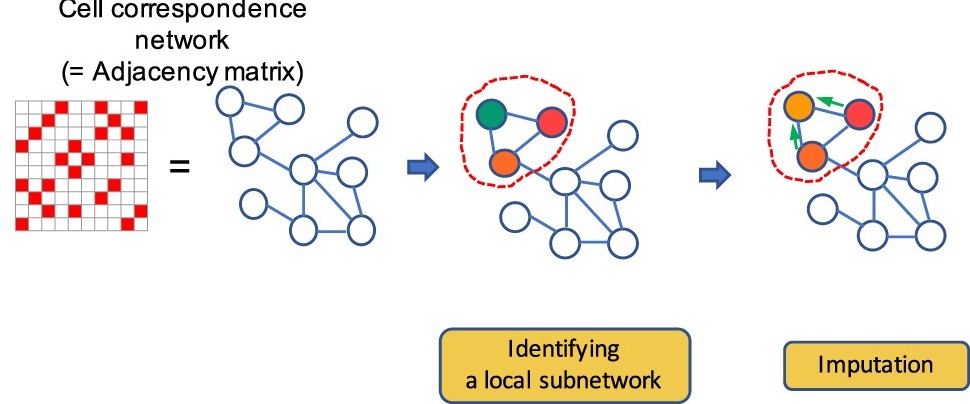
Hyundoo Jeong and Zhandong Liu. “PRIME: a probabilistic imputation method to reduce dropout effects in single cell RNA sequencing.” Bioinformaitcs. doi: 10.1093/bioinformatics/btaa278
Single-cell RNA sequencing technology is vulnerable to a type of noise called dropout effects, which lead to zero-inflated distributions in the transcriptome profile and reduce the reliability of the results. We describe a novel imputation method that reduces dropout effects in Expression profiles of single-cell sequencing, on synthetic and eight real single-cell sequencing datasets and verified that it improves the quality of visualization and accuracy of clustering analysis and can discover gene expression patterns hidden by noise.
2019
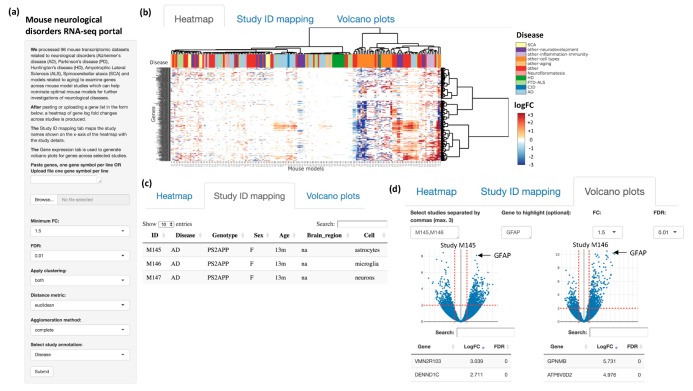
Rami Al-Ouran, Ying-Wooi Wan, Carl Grant Mangleburg, Tom V. Lee, Katherine Allison, Joshua M. Shulman, and Zhandong Liu. “A portal to visualize transcriptome profiles in mouse models of neurological disorders.” Genes 10, no. 10 (2019): 759. doi:10.3390/genes10100759
Target nomination for drug development has been a major challenge in the path to finding a cure for several neurological disorders. Comprehensive transcriptome profiles have revealed brain gene expression changes associated with many neurological disorders, and the functional validation of these changes is a critical next step. To help in nominating the best mouse models for studying neurological diseases, we developed a web portal to visualize mouse transcriptomic data related to neurological disorders

Julia Wang, Zhandong Liu, Hugo J. Bellen, and Shinya Yamamoto. “Navigating MARRVEL, a Web-Based Tool that Integrates Human Genomics and Model Organism Genetics Information.” Journal of visualized experiments: JoVE 150 (2019). doi:10.3791/59542
Through whole-exome/genome sequencing, human geneticists identify rare variants that segregate with disease phenotypes. MARRVEL (Model organism Aggregated Resources for Rare Variant ExpLoration) is a one-stop data collection tool for human genes and variants and their orthologous genes in seven model organisms including in mouse, rat, zebrafish, fruit fly, nematode worm, fission yeast and budding yeast. MARRVEL is an easily accessible open access website designed for both clinical and basic researchers and also serves as starting point to design experiments for functional studies.
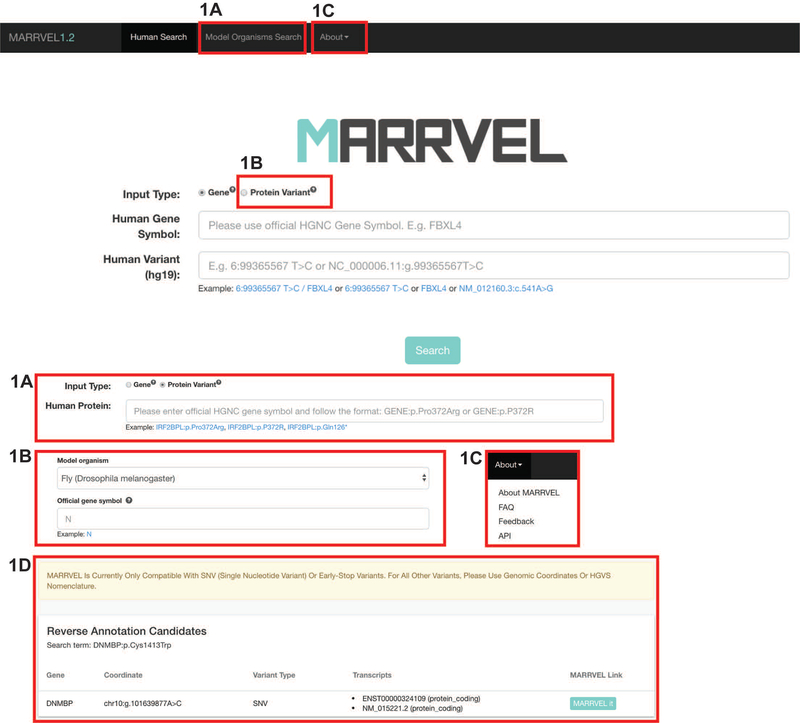
Julia Wang, Dongxue Mao, Fatima Fazal, Seon‐Young Kim, Shinya Yamamoto, Hugo Bellen, and Zhandong Liu. “Using MARRVEL v1. 2 for Bioinformatics Analysis of Human Genes and Variant Pathogenicity.” Current protocols in bioinformatics 67, no. 1 (2019): e85. doi:10.1002/cpbi.85
One of the greatest challenges for bioinformatic analysis of human sequencing data is identifying which variants are pathogenic. To solve this problem, numerous databases and tools have been generated. However, much of these useful data and tools are spread out and requires users to search for their variants of interest through human genetics databases, variant function prediction tools, and model organism databases. To solve this problem, we collected data from and observed human geneticists, clinicians, and model organism researchers to carefully select and display valuable information that facilitates the evaluation of whether or not a variant is likely pathogenic. This program, Model organism Aggregated Resources for Rare Variant ExpLoration (MARRVEL) v1.2 allows users to collect relevant data from 27 public sources for further efficient bioinformatic analysis of human variants for prioritization.
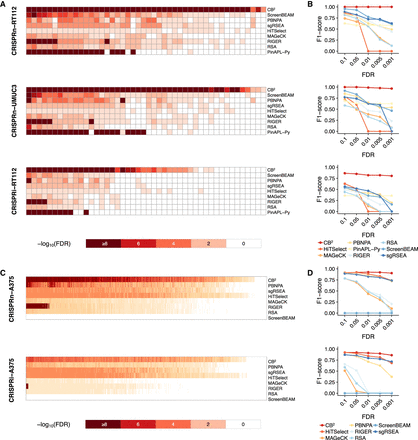
Hyun-Hwan Jeong, Seon Young Kim, Maxime W.C. Rousseaux, Huda Y. Zoghbi, Zhandong Liu. “Beta-binomial modeling of CRISPR pooled screen data identifies target genes with greater sensitivity and fewer false negatives.” Genome Research. doi: 10.1101/gr.245571.118
The simplicity and cost-effectiveness of CRISPR technology have made high-throughput pooled screening approaches accessible to virtually any laboratory. To make CRISPR screen analysis more reliable as well as more readily accessible, we have developed a new algorithm, called CRISPRBetaBinomial or CB2. Based on the beta-binomial distribution, which is better suited to sgRNA data, CB2 outperforms the eight most commonly used methods (HiTSelect, MAGeCK, PBNPA, PinAPL-Py, RIGER, RSA, ScreenBEAM, and sgRSEA) in both accurately quantifying sgRNAs and identifying target genes, with greater sensitivity and a much lower false discovery rate.
2018